Нарушения обмена билирубина.
Билирубин — конечный продукт катаболизма порфиринового кольца молекулы гемоглобина, он не содержит ни железа, ни белка.
Нарушение обмена билирубина связано с расстройством его образования и выделения.
Симптомокомплекс, характеризующийся увеличением количества билирубина в крови с накоплением его в тканях и желтушным окрашиванием кожи, склер, слизистых, серозных оболочек и внутренних органов, называется желтухой.
Желтуха может возникать при наличии следующих условий:
• увеличенное образование билирубина;
• уменьшенная экскреция печенью;
• обструкция желчного протока.
По механизмам развития желтухи различают три ее вида:
• надпеченочную (гемолитическую) — характеризуется повышенным образованием билирубина в связи с увеличенным распадом (гемолизом) эритроцитов;
печеночную (паренхиматозную) — возникает при повреждении гепатоцитов (дистрофии и некрозе их), в результате чего нарушается захват, связывание и экскреция билирубина, что приводит к увеличению его содержания в крови; подпеченочную (механическую) — происходит обтурация желчных путей, что приводит к накоплению связанного билирубина проксимальнее преграды в желчных путях и печени (холестаз).
Гемосидерин— золотисто-желтый, обычно аморфный пигмент, который образуется при расщеплении гема и является полимером ферритина.
Гемосидерин является продуктом внутриклеточного ферментативного расщепления гемоглобина.
Гемосидерин возникает спустя 24 часа от момента кровоизлияния.
Клетки, в которых образуется гемосидерин, называются сидеробластами. В их сидеросомах происходит синтез гранул гемосидерина.
Сидеробласты могут быть как мезенхимальной, так и эпителиальной природы. Гемосидерин постоянно обнаруживается в ретикулярных и эндотелиальных клетках селезенки, печени, костного мозга, лимфатических узлов.
В межклеточном веществе он подвергается фагоцитозу сидерофагами. Присутствие в гемосидерине железа позволяет выявлять его с помощью характерных реакций:
• образование берлинской лазури (реакция Перлса);
• турнбулевой сини (обработка срезов сульфидом аммония);
• железосинеродистым калием и хлористоводородной кислотой.
Положительные реакции на железо отличают гемосидерин от сходных с ним пигментов (гемомеланин, липофусцин, меланин, билирубин).
Избыточное образование гемосидерина в условиях патологии носит название гемосидероза.
Различают общий и местный гемосидероз.
Местный гемосидероз — состояние, развивающееся при вне-сосудистом разрушении эритроцитов (экстраваскулярный гемолиз), т. е. в очагах кровоизлияний.
Местный гемосидероз может возникать в пределах не только участка ткани (гематома), но и целого органа.
Общий, или генерализованный, гемосидероз наблюдается при внутрисосудистом разрушении эритроцитов (интраваскулярный гемолиз).
Причины общего гемосидероза: болезни системы органов кроветворения; интоксикации, обусловленные гемолитическими ядами и солями тяжелых металлов; некоторые инфекционные заболевания; переливания иногруппной, резус несовместимой и бактериально загрязненной крови.
Избыточное железо депонируется как гемосидерин в макрофагах всех органов, особенно костного мозга, печени и селезенки. Роль сидеробластов выполняют в этих органах ретикулярные, эндотелиальные и гистиоцитарные элементы.
Гемосидероз может быть диагностирован в костном мозге и печени при биопсии и не имеет особого клинического значения.
Яркие формы гемосидероза, особенно печени («пигментный цирроз»), поджелудочной железы, слюнных желез, наблюдаются при гемохроматозе.
Гемохроматоз — это своеобразное, близкое к общему гемосидерозу заболевание, главным отличием которого является степень перегрузки железом и наличие повреждений паренхиматозных клеток.
Гемохроматоз может быть первичным (наследственным) и вторичным.
Первичный гемохроматоз — самостоятельное заболевание из группы болезней накопления. Передается доминантно-аутосомным путем и связан с наследственным дефектом ферментов тонкой кишки.
Основными симптомами болезни являются: бронзовая окраска кожи; сахарный диабет; пигментный цирроз печени, ведущий к печеночной недостаточности; пигментная кардиомиопатия, которая может стать причиной смерти.
Вторичный гемохроматоз — заболевание, развивающееся при приобретенной недостаточности ферментных систем, обеспечивающих обмен пищевого железа, что сопровождается генерализованным гемосидерозом.
Причиной этой недостаточности могут быть избыточное поступление железа с пищей (железосодержащие препараты), резекция желудка, хронический алкоголизм, повторные переливания крови и др.
При вторичном гемохроматозе содержание железа повышено не только в тканях, но и в сыворотке крови.
Основные клинико-морфологические проявления заболевания аналогичны тем, что наблюдаются при первичном гемосидерозе.
Не нашли то, что искали? Воспользуйтесь поиском:
источник
Существуют три типа нарушений обмена билирубина
Ситуации, при которых в крови накапливается билирубин, в зависимости от причины делятся на три вида:
- Гемолитические – в результате гемолиза при избыточном превращении гемоглобина в билирубин,
- Печеночно-клеточные – когда печень не в состоянии обезвредить билирубин,
- Механические – если билирубин не может попасть из печени в кишечник из-за механического перекрытия желчевыводящих путей.
В норме билирубин и его фракции находятся в крови в концентрации не более 20 мкмоль/л, но накопление билирубина в крови свыше 43 мкмоль/л ведет к связыванию его эластическими волокнами кожи и конъюнктивы, что проявляется в виде желтухи . Поскольку свободный билирубин липофилен, то он легко накапливается в подкожном жире и нервной ткани. Последнее очень опасно для детей, особенно для новорожденнных, т.к. происходит резкое нарушение окислительного фосфорилирования и образования АТФ в нейронах.
Гемолитическая желтуха
Гемолитическая (надпеченочная) желтуха – ускоренное образование билирубина в результате усиления внутрисосудистого гемолиза любого происхождения – сепсис, лучевое поражение, несовместимость крови по AB0 или резус-фактору, дефект глюкозо-6-фосфатдегидрогеназы пентозофосфатного пути, отравление гемолитическими ядами (хлорбензол, яд кобры), малярия. У новорожденных желтуха может развиться как симптом гемолитической болезни новорожденного.
Гепатоциты усиленно переводят избыток непрямого билирубина в связанную форму, секретируют его в желчь, в результате в кале увеличивается содержание стеркобилина, интенсивно его окрашивая.
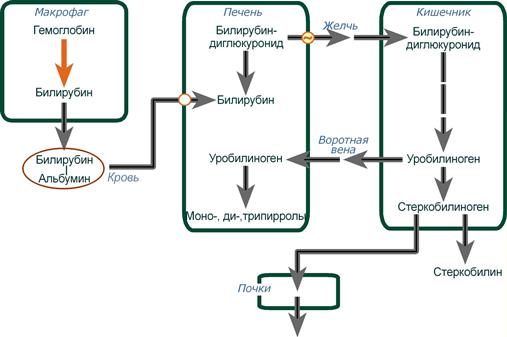
Схема патогенеза гемолитической желтухи
Гемолитические состояния, независимо от причины, имеют характерные проявления – синдром гемолиза . Биохимическими маркерами синдрома являются:
- Гипербилирубинемия за счет повышения содержания в крови свободного (непрямого) билирубина.
- Интенсивная окраска мочи, обусловленная накоплением в ней пигмента стеркобилина (билирубина и уробилина нет).
- Насыщенный цвет кала за счет увеличенного содержания в нем стеркобилина.
- Низкий уровень гаптоглобина в крови.
- Повышение в сыворотке крови активности лактатдегидрогеназы-5 (из эритроцитов).
Механическая желтуха
Механическая (подпеченочная) желтуха развивается вследствие снижения оттока желчи при непроходимости желчного протока (закупорка желчного протока опухолями, желчными камнями). При этом происходит растяжение желчных капилляров, увеличивается проницаемость их стенок и не имеющий оттока в кишечник прямой билирубин поступает в кровь, развивается гипербилирубинемия.
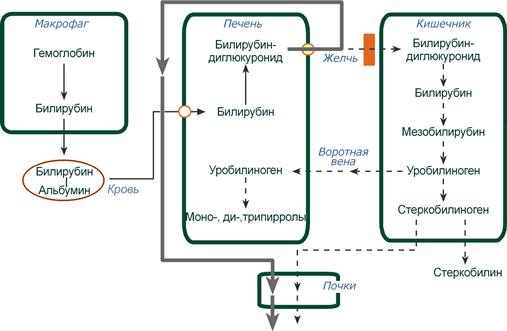
Схема патогенеза механической желтухи
Биохимическими маркерами синдрома холестаза (в «чистом» виде, без повреждения гепатоцитов) являются:
- Гипербилирубинемия за счёт связанного (прямого) билирубина.
- В моче высокий уровень билирубина (коричневый цвет, цвет темного пива) и снижено количество стеркобилина, уробилина нет.
- В кале практически отсутствует стеркобилин (обесцвеченность, серовато-белое окрашивание).
- Повышение в сыворотке крови активности ферментов, специфичных для желчных канальцев – щелочная фосфатаза (желчный изофермент), 5′-нуклеотидаза, γ-глутамилтранспептидаза.
- Уменьшение концентрации альбуминов и увеличение содержания α2-, β- и γ-глобулинов в сыворотке крови (протеинограмма для механической желтухи).
Паренхиматозная желтуха
Паренхиматозная (печеночно-клеточная) желтуха – причиной может быть нарушение на всех трех стадиях превращения билирубина в печени:
- извлечение билирубина из крови печеночными клетками,
- конъюгирование билирубина,
- АТФ-зависимая секреция в желчь.
Наблюдается при различных формах гепатитов (вирусные, токсические) и иных поражениях печени.
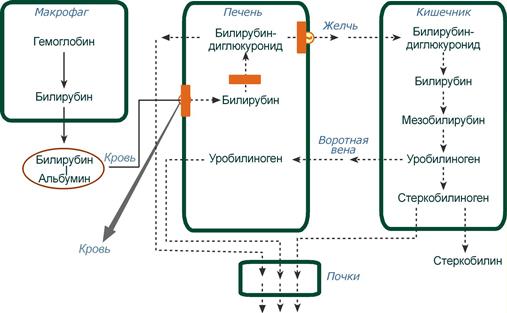
Схема патогенеза паренхиматозной желтухи
У младенцев вариантами паренхиматозной желтухи являются физиологические желтухи новорожденных и недоношенных:
- физиологическая желтуха,
- желтуха, вызываемая молоком матери и т.п.
Наследственные желтухи печеночного происхождения представляют собой синдромы Жильбера-Мейленграхта , Дубина-Джонсона , Криглера-Найяра .
Биохимическими маркерами синдрома цитолиза являются:
- Гипербилирубинемия за счёт обеих фракций билирубина – прямой и непрямой. Их концентрация в крови возрастает из-за одновременного нарушения секреции в желчь и увеличения проницаемости мембран клеток печени.
- В моче имеется билирубин (коричневый цвет, цвет черного чая), появляется уробилин.
- В кале стеркобилин снижен или в норме.
- Повышение в сыворотке крови активности ферментов, специфичных для гепатоцитов – ЛДГ-5, АЛТ, АСТ, γ-глутамилтранспептидаза, глутаматдегидрогеназа.
- Уменьшение концентрации альбуминов и увеличение содержания β- и γ-глобулинов в сыворотке крови (протеинограмма для гепатитов).
источник
19.Эндогенные пигменты. Их виды. Нарушения обмена билирубина

Нарушения обмена билирубина связаны с его образованием и выделением. Билирубин, желчный пигмент, не содержит железа, образуется в печени при восстановлении биливердина, производного порфиринового кольца гема, простетической группы гемоглобина. При нормальных условиях билирубин встречается в желчи и в плазме крови. При патологических состояниях количество билирубина в плазме крови повышается (билирубинемия) и избыток его выделяется с мочой (билирубинурия), кожные покровы, склеры, слизистые и серозные оболочки внутренних органов окрашиваются в желтый цвет.
По механизму образования различают 3 вида желтухи:
1. Надпеченочную (гемолитическая)
2. Печеночную или паренхиматозную.
3. Подпеченочную (механическую).
Надпеченочная желтуха возникает при внутрисосудистом разрушении эритроцитов (гемолиз). Причины, вызывающие гемолитическую желтуху: сепсис, малярия, возвратный тиф и интоксикации (гемолитическими ядами), переливание несовместимой крови. К гемолитической желтухе относятся желтуха новорожденных в первые дни жизни, а также несовместимость резус фактора ребенка и матери.
Печеночная желтуха развивается при поражении гепатоцитов, когда гепатоциты не способны связывать билирубин с глюкуроновой кислотой и нарушается его выведение. Печеночная желтуха возникает при остром и хроническом гепатите, циррозах печени, отравлении фосфором.
Подпеченочная желтуха связана с нарушением проходимости желчных протоков. Эта желтуха наблюдается при желчнокаменной болезни, раке желчных путей, головки поджелудочной железы, раке фатерова сосочка. В печени при застое желчи возникают очаги некроза с последующим замещением соединительной тканью и развитием цирроза.
Накопление билирубина в клетках печени при обтурационой желтухе приводит к токсическому повреждению – дистрофии, и, при тяжелом поражении, некрозу. Затем в участках некроза развивается фиброз, что может привести к билиарному циррозу и хронической печененочной недостаточности. С аутоинтоксикацией связано поражение почек, развитие печеночно-почечной недостаточности.
20.НАРУШЕНИЯ ОБМЕНА ПРОТЕИНОГЕННЫХ (ТИРОЗИН-ТРИПТОФАНОВЫХ) ПИГМЕНТОВ. К протеиногенным (тирозиногенным) пигментам относят: меланин; пигмент гранул энтерохромаффинных клеток; адренохром.
Меланин черно-бурый пигмент. Синтез меланина происходит из тирозина в клетках меланинобразующей ткани – меланоцитах, имеющих нейроэктодермальное происхождение. Различают приобретенный и врожденный меланоз. Он может быть распространенным и локализованным.-Распространенный приобретенный гипермеланоз в клинике проявляется в виде гиперпигментации кожи.
Причины распространенного приобретенного гипермеланоза:
поражение надпочечников туберкулезной или опухолевой природы (адиссоновая болезнь), амилоидоз; эндокринные расстройства (гипогонадизм, гипопитуитаризм);
авитаминозы (пеллагра, цинга); интоксикации углеводородами.
Значение процесса определяется тяжестью основного заболевания.
-Распространенный врожденный гипермеланоз (пигментная ксеродерма) характеризуется повышенной чувствительностью кожи к ультрафиолетовым лучам и выражается в пятнистой пигментации кожи с явлениями гиперкератоза и отека.
-Очаговый приобретенный гипермеланоз. Примеры:
меланоз толстой кишки (у людей, страдающих хроническими запорами); пигментные пятна кожи (веснушки (эфелиды), лентиго); очаговая гиперпигментация при аденомах гипофиза, гипертиреоидизме, сахарном диабете, пигментные невусы, меланомы.
АДДИСОНОВА БОЛЕЗНЬ, обусловленное разрушением надпочечников или снижением их функции в результате дефицитом гормонов этих желез. Причины. К разрушению ткани, чаще всего туберкулез., главной причиной является «ошибка» иммунной системы, /Основное значение при аддисоновой болезни имеет недостаточность гормонов кортизола и альдостерона. Признаки и симптомы. Наиболее яркий признак аддисоновой болезни – прогрессирующее потемнение кожи, обусловленное избыточной продукцией кожного пигмента меланина. Причиной усиленного образования меланина служит АКТГ (адренокортикотропный гормон), вырабатываемый гипофизом. В норме секреция кортизола регулируется системой «обратной связи» между гипофизом и надпочечниками: как только уровень кортизола повышается, наступает торможение секреции АКТГ, что снова ведет к падению концентрации кортизола – и цикл повторяется. Поскольку при аддисоновой болезни надпочечники неспособны повысить продукцию кортизола, секреция АКТГ выходит из-под контроля, а его повышенный уровень в крови приводит к увеличению пигментации кожи. Другим частым признаком аддисоновой болезни является сильное похудание.
21) НАРУШЕНИЕ ОБМЕНА ПРОТЕИНОГЕННЫХ ПИГМЕНТОВ. ОСЛАБЛЕНИЕ ПИГМЕНТАЦИИ: РАСПРОСТРАНЕННОЕ И МЕСТНОЕ, ПРИОБРЕТЕННОЕ И ВРОЖДЕННОЕ. АЛЬБИНИЗМ. НАРУШЕНИЕ ОБМЕНА ЛИПИДОГЕННЫХ ПИГМЕНТОВ. ЛИПОФУСЦИНОЗ. ПРИЧИНЫ, МОРФОЛОГИЧЕСКАЯ ХАРАКТЕРИСТИКА, ИСХОДЫ.
Распространенный гипомеланоз: развивается при эндокринных расстройствах (гипогонадизм, гипопитуитаризм), когда угнетена регуляция синтеза меланина.
Первичный распространенный гипомеланоз называется альбинизмом: заболевание обусловлено отсутствием (полный альбинизм) или уменьшением (частичный альбинизм) фермента тирозиназы. Меланин отсутствует в волосяных луковицах, эпидермисе и дерме, в сетчатке и радужной оболочке глаз. Поэтому у лиц с этой патологией белая кожа, бесцветные волосы, красная радужная оболочка глаз, а также выраженная фотобоязнь (фотофобия), блефароспазм, ожоги кожи при инсоляции.
Местные гипомеланозы: относят очаговые депигментированные участки на коже, которые называются в и т и л и г о, или лейкодерма. Они возникают на коже в результате действия некоторых лекарственных (фурацилин) и химических (синтетические смолы) веществ, нервно-трофических (лепра, сифилис), нейроэндокринных (СД, гипопаратиреоз) и аутоиммунных (зоб Хасимото) факторов меланогенеза, а также после воспалительных и некротических процессов на коже. При вторичном рецидивном сифилисе, например, описывают «ожерелье Венеры» .
Нарушения обмена липидогенных пигментов: В эту группу входят жиро-белковые пигменты — липофусцин, гемофусцин, пигмент недостаточности витамина Е, цероид и липохромы.
Липофусцин представляет собой гликопротеид. Светооптически он представлен зернами золотистого или коричневого цвета в цитоплазме клеток печени, почек, миокарда, скелетных и гладких мышц, симпатических ганглиев и коры надпочечников. Электронно-микроскопически пигмент выявляется в виде электронно-плотных гранул, окруженных трехконтурной мембраной. При язвенной болезни желудка и 12-ти перстной кишки развивается липофусциноз печени, при пороке сердца — липофусциноз миокарда.
→нарушения обмена липофусцина выражается в избыточном его накоплении — липофусцинозе, который может быть первичным (наследственным) и вторичным.
Первичный (наследственный) липофусциноз: характеризуется избирательным накоплением липофусцина в клетках органа или системы (чаще встречаются с поражением ЦНС). Накопление липофусцина в клетках ЦНС наблюдают при нейрональных липофусцинозах (болезнь Тея — Сакса). Морфология: в различных отделах и клетках НС обнаруживают избыточное скопление липофусцина, баллонную дистрофию, распад нервных клеток, в тяжелых случаях — демиелинизацию и разрушение аксонов, что носит вторичный характер в связи с накоплением липофусцина.
Клиника: проявляются снижением интеллекта вплоть до идиотии, двигательными расстройствами (судороги, параличи), расстройствами зрения до полной слепоты. Если проявляется в раннем детском возрасте (болезнь Бильшовского — Янского) или в возрасте 6—10 лет (юношеская форма Баттена — Шпильмейера — Фогта), то быстро прогрессирует и заканчивается смертью на фоне выраженной идиотии. У взрослых процесс затягивается на 10—15 лет, развиваются параличи, припадки и органические изменения психики; слепота не возникает, но прогноз тоже фатальный.
Накопление липофусцина возможно в печени. В этих случаях развивается пигментный гепатоз, или доброкачественная гипербилирубинемия (генетически обусловленной недостаточностью ферментов, обеспечивающих захват и глюкуронизацию билирубина в гепатоцитах).
Проявления: нарушение пигментного обмена выражается в преходящей желтухе, все остальные функции печеночной клетки не страдают. Различают пигментные гепатозы с конъюгированной и неконъюгированной гипербилирубинемией, причиной некоторых является недостаточность ферментов, например глюкуронилтрансферазы.
Вторичный липофусциноз: развивается при гипоксии, когда увеличивается потребность в кислороде, в старости и при истощающих заболеваниях, когда выражены нарушения окислительных процессов и отсутствуют антиоксиданты, снижающие потребность тканей в кислороде. В этих случаях паренхиматозные органы уменьшаются в размерах, в них прогрессирует склероз, который усугубляется гипоксией, и липофусциноз — развивается бурая атрофия печени, миокарда, поперечнополосатой мускулатуры. При кахексии (алиментарной церебральной и др.) нарушается синтез окислительно-восстановительных ферментов в цепи цитохромов, метаболизм клеток переключается на более «экономный» липофусциновый путь — развивается бурая атрофия органов.
22.НАРУШЕНИЯ ОБМЕНА НУКЛЕОПРОТЕИДОВ Конечным продуктом обмена нуклеопротеидов является мочевая кислота и ее соли. В норме мочевая кислота выводится почками и кишечником. При нарушении обмена нуклеопротеидов и избыточном образовании мочевой кислоты соли откладываются в тканях, что наблюдается при подагре, мочекислом инфаркте и мочекаменной болезни.
Подагра — хроническое заболевание, характеризуется нарушением обмена веществ и отложением мочевой кислоты и ее солей в тканях: в области суставов, чаще в тканях мелких суставов ног и рук, в сухожилиях и суставных сумках, — в хряще ушных раковин, — в почках. Ткани, в которых выпадают соли в виде кристаллов или аморфных масс, некротизируются. Вокруг отложения солей и очагов некроза, развивается воспалительная реакция со скоплением гигантских клеток. По мере увеличения отложений солей и разрастания вокруг них соединительной ткани образуются подагрические шишки (tophi urici), суставы деформируются. В почках соли мочевой кислоты скапливаются в канальцах и собирательных трубках.
.-Первичная подагра обусловлена врожденными нарушениями пуринового обмена. Об этом свидетельствует ее семейный характер и сочетание подагры с другими нарушениями обмена веществ (ожирение, диабет, желчекаменная болезнь). Велика роль в развитии заболевания особенностей питания, употребления больших количеств животных белков, сухих вин, шампанского, пива, неподвижного образа жизни. Соли обычно выпадают в синовии и хрящах мелких суставов, в сухожилиях и суставных сумках, в хряще ушных раковин. В почках отмечается накопление мочевой кислоты в канальцах с обтурацией их просвета, развитие вторичных воспалительных и атрофических изменений -подагрические почки.
—Вторичная подагра является осложнением: опухолей кроветворной ткани (при усиленном распаде опухолевых клеток); эндокринных заболеваний; болезней почек различной этиологии с исходом в нефроцирроз.
Мочекаменная болезнь, как и подагра, может быть связана прежде всего с нарушениями пуринового обмена, т.е. быть проявлением так называемого мочекислого диатеза. В почках при этом образуются исключительно или преимущественно ураты.
Мочекислый инфаркт встречается у новорожденных, проживших не менее двух суток, и проявляется выпадением в канальцах и собирательных трубках почек аморфных масс мочекислых натрия и аммония. Макроскопически они видны в виде желто-красных полос сходящихся у сосочков мозгового слоя почки. Возникновение мочекислого инфаркта связано с интенсивным обменом в первые дни жизни новорожденного и отражает адаптацию почек к новым условиям существования.
источник