62.Наследственные гипербилирубинемии. Диагностика желтухи. Нарушения обменов порфиринов.
Наследственные гипербилирубинемии — группа наследственных энзимопатий, характеризующихся нарушением обмена билирубина и проявляющихся стойкой или перемежающейся желтухой при отсутствии выраженных изменений структуры и функции печени, явных признаков гемолиза и холестаза. Заболевания обусловлены нарушением процессов захватывания гепатоцитами свободного билирубина из крови, связывания его с глюкуроновой кислотой с образованием билирубин-глюкуронида (связанного билирубина) и последующего выделения его с желчью.
Виды наследственных гипербилирубинемий:
Синдром Жильбера (ювенильная интермиттирующая желтуха, синдром Мейленграхта) развивается вследствие снижения способности клеток печени захватывать и связывать билирубин. Характеризуется умеренным интермиттирующим повышением содержания несвязанного (непрямого) билирубина в крови, отсутствием других функциональных или морфологических изменений печени и аутосомно-доминантным типом наследования. Основное проявление болезни – желтушность склер, реже кожных покровов, появляющаяся в возрасте 20 лет и старше.
Синдром Криглера-Найяра I типа характеризуется крайне высоким содержанием в крови свободного (непрямого) билирубина из-за отсутствия в гепатоцитах глюкуронилтрансферазы, переводящей свободный билирубин в связанный, токсическим действием билирубина на базальные и стволовые ядра головного мозга с возникающей вследствие этого энцефалопатией, нередко приводящей больных к смерти в детском возрасте, и аутосомно-рецессивным типом наследования. Неврологические проявления включают повышение мышечного тонуса, нистагм, опистотонус, атетоз, тонические и клонические судороги; характерно отставание детей в психическом и умственном развитии.
Синдром Криглера-Найяра II типа обусловлен частичным дефицитом того же фермента, желтуха менее интенсивна, неврологические нарушения не выражены; тип наследования — аутосомно-доминантный. Синдром Дубина-Джонсона (хроническая идиопатическая желтуха) характеризуется умеренным повышением содержания в крови связанного (прямого) билирубина вследствие нарушения механизмов его транспорта из микросом гепатоцитов в желчь, извращенными результатами теста на задержку бромсульфалеина со вторичным повышением его содержания в крови через 90 мин после в/в введения, неконтрастированием желчного пузыря при пероральной холецистографии, черным цветом печени из-за скопления в гепатоцитах коричневого липохромного пигмента, повышенным выделением с мочой копропорфирина и аутосомно-рецессивным типом наследования.
Ротора синдром (гипербилирубинемия идиопатическая типа Ротора) характеризуется умеренным повышением содержания в крови связанного билирубина, задержкой выделения печенью введенного в/в бромсульфалеина, повышенным выделением почками копропорфирина, отсутствием других функциональных и морфологических изменений печени, аутосомно-рецессивным типом наследования.
Симптомы наследственных гипербилирубинемий:
Во всех случаях гипербилирубинемия и желтуха обнаруживаются с раннего детства, в большинстве случаев (кроме синдрома Криглера-Найяра) – незначительны, могут иметь перемежающийся характер (усиливаться под влиянием погрешностей в диете, приема алкоголя, интеркуррентных заболеваний, физического переутомления и других причин).
Нередки нерезко выраженные диспептические явления, легкая астенизация, слабость, быстрая утомляемость. Печень обычно не увеличена, мягка, безболезненна, функциональные пробы печени (за исключением гипербилирубинемии) не изменены. Радионуклидная гепатография изменений не выявляет. Селезенка не увеличена. Осмотическая резистентность эритроцитов, продолжительность их жизни – нормальны. Пункционная биопсия при всех формах (кроме синдрома Дубина-Джонсона) изменений не выявляет.
Течение при всех формах (кроме синдрома Криглера-Найяра) доброкачественное, прогноз благоприятный; трудоспособность, как правило, сохранена. При синдроме Криглера-Найяра I типа больные обычно погибают в раннем детском возрасте, при II типе пациенты обычно доживают до зрелого возраста.

Нарушения обменов порфиринов:
Порфирии — группа заболеваний (преимущественно наследственных), при которых частичный дефицит одного из ферментов синтеза порфирина приводит к уменьшению образования гемма и, следовательно, к устранению подавляющего эффекта гемма на АЛК-синтазу, результатом чего является образование избыточных количеств предшественников порфиринов и порфиринов.
Клиника зависит от накопления тех или иных промежуточных продуктов.
Образование избыточных количеств ранних предшественников порфирина — ДАЛК, порфобилиногена (нейротоксины) →неврологические расстройства.
Накопление порфиринов — воспалительная реакция в ответ на освещение кожных покровов светом. Механизм — порфирины абсорбируют свет,переходят в возбужденное состояние, вызывая образование токсичных свободных радикалов).
Для подтверждения диагноза необходимо провести специальное обследование, включающее исследование мочи на содержание порфобилиногена (при порфирии его количество повышается), определение активности порфобилиноген дезаминазы и мутаций в гене порфобилиноген дезаминазы (в гене PBGD).
Для исключения кожных порфирий определяют содержание порфиринов в эритроцитах, плазме крови, моче и кале, оценивают спектр поглощения порфиринов при помощи флюоресцентной спектроскопии. Проводят также определение содержания порфобилиногена в моче и ДАЛК в моче.
Основная часть синтезируемых порфиринов эритробластов идет на образование Нв, а неиспользованные порфирины и их предшественники выделяются с мочой и калом.
Различные типы порфиринов (уро-копро-прото) образуются в зависимости от того, какими радикалами замещены атомы водорода в порфине.Названы по источнику первоначального выделения.
Порфирины в моче — преимущественно копропорфирины >уропорфирины > порфобилиноген и ДАЛК. Основной источник – печень, меньше эритронормобласты.
Порфирины в кале — копропорфирины и протопорфирины. Часть порфиринов в организме экзогенного происхождения(поступают с пищей в составе мясных продуктов, овощей, фруктов). В печени они превращаются в копропорфирины и выделяются с желчью.
источник
10. Нарушение обмена билирубина. Желтуха, ее виды и их характеристика. Наследственные гипербилирубинемии.
обычно связано с нарушением его образования и выделения, что ведет к накоплению его в крови- билирубинемии и обусловливает окраску склер, кожи, слизистых в желтый цвет. Поэтому такое состояние обозначается термином желтуха.
а\ Гемолитическая\ надпеченочная— Эта желтуха развивается при повышенном распаде эритроцитов в клетках и образовании большого количества непрямого билирубина. Желтуха, как результат билирубинемии, развивается потому, что печень не успевает переработать весь непрямой билирубин. Проявления патологии- Кровь – много непрямого билирубина.Кал – интенсивно окрашен в желтый цвет, в нем много стеркобилина.Моча — много уробилина.
б\Паренхиматозная \ печеночная\ в результате нарушения захвата билирубина и его конъюгации с глюкуроновой кислотой и экскреции. Отмечается при повреждении гепатоцитов и , следовательно, снижении функции печени , отчего даже обычное количество внепеченочного билирубина не перерабатывается прямой билирубин. Проявления патологии-Кал – бледно окрашен\ мало желчи\ и мало стеркобилина.Моча — мало уробилина. Кровь – увеличение непрямого билирубина. Паренхиматозная желтуха часто отмечается при вирусных гепатитах ; гепатозах – токсических повреждениях печени ядовитыми грибами, медикаментами, солями тяжелых металлов, четыреххлористым углеродом, при беременности и энзимопатиях.
в\ Механическая желтуха Возникает при наличии препятствия для прохождения желчи в желчевыводящих протоках. Затруднение экскреции и регургитация желчи. Причины:1\ камень, закупоривающий общий желчный проток, опухоль поджелудочной железы. При застое желчи в печени возникают очаги некроза с последюущим их замещением ст и развитием цирроза (вторичный билиарный цирроз). Ведет к холемии.
Проявления желтухи1\ Яркое интенсивное желтое окрашивание с зеленоватым оттенком кожи и слизистых 2\ Бесцветный кал цвета известковой замазки \ отсутствие желчи\, в кале отсутствует стеркобилин3\ В моче нет уробилина 4\ Кровь — большое количество прямого билирубина, желчных кислот. Накопление в крови прямого билирубина и желчных кислот предопределяет тяжелую общую интоксикацию и повреждение всех органов и тканей — центральной нервной системы, сердца, почек . Х-м проявлением механической желтухи являются приступы нестерпимого кожного зуда вследствие раздражения нервных рецепторов желчными кислотами.
Среди неконъюгированных гипербилирубинемий основную массу составляют болезнь Жильбера и примыкающие к ней синдромы. Остальные гипербилирубинемий встречаются очень редко (синдромы Криглера — Найяра, Дубина — Джонсона, Ротора). Точных статистических данных о частоте этих синдромов нет.
Болезнь Жильбера относится к наследственным заболеваниям, передающимся по аутосомно-доминантному типу. В патогенезе заболевания играют роль, во-первых, нарушения транспортной функции белков (глутатионтрансферазы и др.), доставляющих неконъюгирован-ный билирубин к гладкому эндоплазматическому ретикулуму (микросомам) гепатоцита и, во-вторых, неполноценность фермента микросом УДФ-глюкуронилтрансферазы, при помощи которого осуществляется конъюгация (соединение) билирубина с глюкуроновой и другими кислотами.
Нарушение конъюгации непрямого билирубина- Криглера- Найяра до ядерной желтухи
Нарущение выведения прямого билирубина- синдром Дабина-Джонсона, Ротора.
11.Нарушение обмена Кальция.Метаболизм кальция в организме. Кальцинозы- причины, патогенез,виды, морфологическая характеристика
Влияет на 1\функцию клеточных мембран, 2\возбудимость нервно-мышечного аппарата 3\ свертывание крови 4\ кислотно-щелочное равновесие 5\ формирование скелета .

Кальциноз выражаются выпадением кальция в тканях с образованием структур каменистой плотности. Матрицей для кальция могут быть белки, гликопротеиды, эластические и коллагеновые волокна.
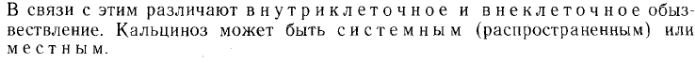
1. Различают 3 типа известковых дистрофий: 1\метастатическое 2\ дистрофическое 3\ метаболическое
1\ Метастатические — отмечается при интенсивном вымывании кальция из костной ткани и насыщение им крови и тканевой жидкости.
Массивное вымывание кальция из костей отмечается –а\ при гиперпродукции паратгормона в случаях аденомы и гиперплазии паращитовидной железы. В итоге развивается фиброзная остеодистрофия, кости становятся гибкими наподобии резиновых жгутиков. б\ при разрушении костей метастазами опухолей\ рак легких, рак щитовидной железы\ в\ разрушении костей при миеломной болезни\ опухоль из плазматических клеток\ Места отложения кальция- легкие, желудок, стенки артерий, почки, миокард, где отмечается щелочная среда и уменьшение углекислоты. В местах скопления извести, которые легко определяются и макроскопически и микроскопически часто развивается воспалительная реакция.
2\ Дистрофическое обызвествление,петрификация наблюдается в местах глубокой дистрофии и некроза, имеет местный характер. На местах дистрофического обызвествления образуются каменистые образования. А в дальнейшем формируется костная ткань. Петрификация и оссификация являются признаками благоприятного исхода на местах туберкулезных, сифилитических очагов, зон некрозов, хронического воспаления, погибших паразитов, мертвого плода при внематочной беременности.
3\ Метаболическое обызвествление известковая подагра, интерстициальный кальциноз развивается в местах без предшествующего патологического процесса, первично. Это связано с нестойкостью буферных систем, неспособностью коллоидов и белков удерживать кальций в растворенном состоянии.
Исход- неблагоприятен, выпавшая известь не рассасывается или с трудом
источник
Наследственные нарушения обмена билирубина (наследственные гипербилирубинемии)
Встречается несколько форм наследственной гипербилирубинемии, сопровождающейся желтухой различной степени.
Наиболее легкая форма наследственной гипербилирубинемии (конституциональная дисфункция печени, асимптоматическая семейная, неконъюгированная доброкачественная гипербилирубинемия).
В печени патологические изменения незначительны и представлены увеличением содержания липофусцина, главным образом в центролобулярных гепатоцитах. При электронно-микроскопическом исследовании [McGee Л. et al., 1975; Dawson J. et al., 1979; Lapis K., 1979] в части случаев находят гиперплазию гладкой эндоплазматической сети, которая может быть ошибочно расценена как проявление тяжелого заболевания гепатобилиарной системы.
Различают два самостоятельных типа синдрома. I тип наследуется аутосомно-рецессивно и проявляется только у гомозигот. Его основу составляет полное или частичное отсутствие глюкуронилтрансферазы. что сопровождается тяжелой степенью неконъюгированной гипербилирубинемии. Возникают тяжелые изменения головного мозга (поражение ядер), которые приводят к смерти больных в раннем младенческом возрасте [Huang Р. еt al., 1970]. Конъюгированный билирубин выводится с желчью в ничтожном количестве, поэтому желчь в таких случаях почти бесцветная. В печени наблюдается каналикулярный холестаз. иногда желчный пигмент находят и в ЗРЭ [Huang Р. et al., 1970]. Экспериментальной моделью этого типа синдрома Криглера — Найяра являются крысы линии Gunn, страдающие тяжелой недостаточностью глюкуронилтраисферазы.
II тип синдрома наследуется аутосомно-доминантно и характеризуется лишь легкой степенью недостаточности глюкуронилтрансферазы. В связи с этим некопъюгированная гипербилирубинемия представлена умеренно, желтушное поражение ядер мозга развивается редко [Gordon Е. et al., 1976]. Изменения печени подобны таковым при I типе синдрома.
Встречается крайне редко, представляет группу симптомов, развивающихся в связи с наследственным дефектом выделения билирубина гепатоцитами. Другие фазы билирубинового обмена (захват и конъюгация) не нарушены, поэтому результатом дефекта выделения является конъюгированная гипербилирубинемия. При этом синдроме отсутствует выделение не только билирубина, но и других органических анионов (например, бромсульфалеина), а также контрастных веществ, используемых для холецистографии (желчный пузырь при холецистографии не контрастируется).
Кожа у больных желтого циста, печень — темно-коричневого или зеленовато-черного. Пигмент, накапливающийся в увеличенных лизосомах гепатоцитов, имеет форму липофусциновых зерен, однако цвет их темнее и размер больше (рис. 73, а). Эти зерна концентрируются на билиарном полюсе гепатоцитов (рис. 73. б) [Baba N.. Ruppert R., 1972; Swartz И. et al., 1979; Lapis К.. 1979]. Данные пигментные зерна состоят не из били рубина; возможно это липофусцин или меланин, но окончательно природа зерен остается невыясненной. Помимо этих находок, гистологическое строение печени нормально, холестаз отсутствует, поскольку выделение желчи и ее отток не нарушены. После обнаружения недостаточности супероксиддисмутазы у части таких больных Т. Peters и С. Seymour (1978) считают что пигментные зерна соответствуют вторичным лизосомам, со держащим продукты пероксидации липидов клеточной мембраны, или же остаточным тельцам.

73. Печень при синдроме Дабина Джонсона.
а — в гепатоцитах грубые пигментные зерна. ШИК-реакция по Ормос. Х325. б — электронно-плотные пигментные включения с гомогенной структурой. Х-4000.

Семенное заболевание, встречающееся редко и напоминающее синдром Дабина—Джонсона. В основе лежит также дефект выделения билирубина, однако в отличие от синдрома Дабина — Джонсона патологическая пигментация печени отсутствует. Имеются различия между этими синдромами и в отношении нарушений порфиринового обмена [Evans J. еt al., 1981].
источник
Нарушения обмена билирубина, желтуха

Нарушения обмена билирубина, желтуха, гипербилирубемия у детей
Гипербилирубинемии — патологические состояния, характеризующиеся нарушением равновесия между образованием и выделением билирубина, основным клиническим признаком которых является желтуха (иктеричность) — желтая пигментация кожи или склер билирубином, обусловленная повышением содержания общего билирубина в сыворотке крови.
Хотя точный уровень билирубина в крови, при котором желтуха становится клинически явной, варьирует, ее легко можно обнаружить при уровне билирубина выше 34,2 мкмоль/л (норма 8,5—20,5 мкмоль/л). Анализ крови на биохимию, детская поликлиника «Маркушка».
Желтуха чаще диагностируется при потемнении мочи или желтом окрашивании кожи или склер
Желтуха чаще диагностируется при потемнении мочи или желтом окрашивании кожи или склер. Желтая окраска склер объясняется большим количеством в этой ткани эластина, обладающего особым сродством к билирубину, поэтому именно по иктеричности склер часто определяется желтуха.
При выраженной желтухе кожа может приобрести зеленоватый оттенок из-за превращения билирубина в биливердин, продукт окисления билирубина.
Желтуху следует отличать от желтой пигментации кожи (ложной желтухи)
Желтуху следует отличать от желтой пигментации кожи (ложной желтухи), вызываемой другими причинами, такими как каротинемия (при избыточном употреблении пищи, содержащей каротин: тыквы, моркови и красного перца, в особенности при повреждении печени, когда каротин не может быть переработан в витамин А), которая сопровождается желтоватым окрашиванием кожи, но не склер и слизистых оболочек.
Гипербилирубинемия у детей
Гипербилирубинемия возникает вследствие: повышения продукции пигмента; снижения поглощения билирубина печенью; нарушения конъюгации билирубина; снижения экскреции конъюгированного пигмента из печени в желчь.
Надпеченочная (гемолитическая) желтуха
Надпеченочная (гемолитическая) желтуха возникает в результате усиленного гемолиза эритроцитов, обусловленного внутри- или внеэритроцитарными факторами: гемолитические анемии, дефекты эритроцитарных ферментов, гемолитическая болезнь.
Кожа при этом обычно лимонно-желтого оттенка (больные скорее бледны, чем желтушны). Помимо проявлений, обусловленных гемолизом эритроцитов (анемия, ретикулоцитоз, гемоглобинурия), отмечается повышение в крови содержания непрямого билирубина.
Гипербилирубинемия, обусловленная непрямым билирубином
Гипербилирубинемия, обусловленная непрямым билирубином, наблюдается также при нарушении транспорта билирубина (без усиленного гемолиза). Процесс поглощения клеткой печени билирубина включает в себя отщепление пигмента от альбумина и последующее связывание его с лигандином. Уменьшение накопления неконъюгированного билирубина в цитозоле из-за снижения активности Y- и Z-протеинов (конкурентное ингибирование, лихорадочное состояние) приводит к накоплению неконъюгированного билирубина в крови. Анализ крови ребенка — детский медицинский центр «Маркушка».
У некоторых новорожденных, вскармливаемых грудью, развивается выраженная желтуха — синдром Ариаса — за счет накопления в крови непрямого билирубина, уровень которого прогрессивно повышается до 4-го дня жизни и достигает максимума к 10—15-му дню (до 250—300 мкмоль/л), а затем медленно снижается до нормы к 3—12-й неделе жизни.
Гипербилирубинемия выше 12 мкмоль/л встречается у 30 % новорожденных, вскармливаемых грудью. Причиной ее возникновения может быть повышенная активность р-глюкуронидазы грудного молока, вызывающая повышение содержания неконъюгированного билирубина в кишечнике с его последующим всасыванием. Если вскармливание грудью на какой-то период прекратить, то уровень билирубина снижается до нормы в ближайшие 4-8 дн.
Транзиторная семейная гипербилирубемия новорожденных
Транзиторная семейная гипербилирубемия новорожденных отмечается в некоторых семьях, проявляется массивной гипербилирубинемией, развивающейся у всех детей, рожденных от одной матери, страдающей этим заболеванием, в первые 4 года их жизни. Желтуха при этом более интенсивная, сохраняется дольше, чем физиологическая, и связана с наличием ингибирующих субстанций стероидной природы в плазме и моче матери и новорожденного.
Интермиттирующая юношеская желтуха
Интермиттирующая желтуха, чаще возникающая в пубертатном периоде и имеющая доброкачественное течение. Билирубинемия выражена в умеренной степени (уровень билирубина в пределах 17—85 мкмоль/л) и не сопровождается нарушением биохимических показателей функции печени и ее гистологической картины.
Гипербилирубинемия, связанная с нарушением конъюгации билирубина
Гипербилирубинемия, связанная с нарушением конъюгации билирубина (снижение активности билирубинглюкоронилтрансферазы). Почти у каждого новорожденного на 2—5-й день жизни определяется незначительная преходящая неконъюгированная билирубинемия (не выше 150 мг/л) – «физиологическая» желтуха. Она обусловлена возрастной незрелостью глюкоронилтрансферазной системы и исчезает обычно к 7-10-му дню. Степень желтушности у недоношенных детей обычно более значительна, держится дольше (до 4 нед.). Повышение концентрации билирубина может достигать более 200 мкмоль/л, что создает опасность поражения мозга (билирубиновая энцефалопатия).
Длительная и значительно выраженная желтуха при врожденном гипотиреозе
Длительная и значительно выраженная желтуха (до 2-4 мес) наблюдается при врожденном гипотиреозе. У девочек гипотиреоз встречается в 3 раза чаще, чем у мальчиков. Гипотиреоз препятствует нормальному созреванию глюкоронилтрансферазы, уровень билирубина повышается до 220-340 мкмоль/л.
Врожденные и приобретенные нарушения связывания билирубина
Могут встречаться также как врожденные, так и приобретенные нарушения связывания билирубина, обусловленные нарушением активности глюкоронилтрансферазы (синдром Криглера—Найяра, ингибирование глюкуронизации лекарственными средствами).
Приобретенные нарушения активности глюкоронилтрансферазы могут быть вызваны действием лекарственных средств (например, угнетение фермента) или заболеванием печени. Однако при повреждении клеток печени экскреторная способность печени нарушается в большей степени, чем ее способность к связыванию билирубина глюкуроновой кислотой. Поэтому при большинстве гепатоцеллюлярных заболеваний гипербилирубинемия связана в основном с конъюгированным билирубином.
Гипербилирубинемия на фоне преобладания в крови прямого билирубина
Нарушение экскреции билирубина в желчные протоки, независимо от того, обусловлено оно механическими или функциональными факторами, приводит к преимущественному развитию гипербилирубинемии и билирубинурии, связанной с повышением уровня прямого билирубина. Билирубин в моче является важнейшим признаком гипербилирубинемии, связанной с прямым билирубином.
Наследственная конъюгированная гипербилирубинемия
Наследственная конъюгированная гипербилирубинемия проявляется умеренно выраженной желтухой, наследуемой по аутосомно-рецессивному типу. При ней нарушается транспорт билирубина и других органических анионов из печени в желчь. Заболевание проявляется с 2-летнего возраста.
При гепатоцеллюлярном заболевании (гепатит, цирроз) нарушаются три основных этапа обмена билирубина
При гепатоцеллюлярном заболевании (гепатит, цирроз) нарушаются три основных этапа обмена билирубина — его поглощение печенью, связывание и экскреция. Причем экскреция представляет собой этап, ограничивающий скорость обмена билирубина, и обычно именно он нарушается в большей степени. В результате при этих заболеваниях преобладает гипербилирубинемия, обусловленная главным образом прямым билирубином. Вакцинация детей от гепатита В в детской поликлинике «Маркушка».
Паренхиматозная желтуха
Паренхиматозная желтуха чаще возникает при остром вирусном и токсическом гепатитах, хроническом гепатите, циррозе печени, реже — при вторичных гепатитах, развивающихся при различных инфекционных заболеваниях (инфекционном мононуклеозе, инфекции Коксаки, лептоспирозе), бактериальных заболеваниях (сепсисе, тифе, бруцеллезе и др.).
При повреждении печеночных клеток возникает сообщение между желчными путями, кровеносными и лимфатическими сосудами, через которое желчь поступает в кровь и частично в желчевыводящие пути. Отек перипортального пространства также может способствовать обратному всасыванию желчи из желчных ходов в кровь. Набухшие печеночные клетки сдавливают желчные протоки, создавая механическое затруднение оттоку желчи. Нарушаются метаболизм и функции печеночных клеток. Билирубинемия обусловлена преимущественно конъюгированным билирубином. В то же время в крови повышается уровень свободного билирубина, что связано со снижением таких функций гепатоцита, как захват, внутриклеточный транспорт свободного билирубина и его связывание в глюкурониды. Попадание в кровь вместе с желчью желчных кислот обусловливает развитие холемического синдрома. Уменьшение поступления желчи в кишечник (гипохолия, ахолия) ведет к понижению образования метаболитов билирубина и их выделения с мочой и калом (следы стеркобилина), а также появлению симптомов ахолического синдрома. Насыщенный желтый цвет мочи обусловлен повышенным содержанием в ней конъюгированного билирубина (билирубинурия) и уробилина, который недостаточно разрушается в печени после поступления в нее благодаря печеночно-кишечному кругообороту, попадает в общий кровоток и выводится почками (уробилинурия).
Желтуха наблюдается при нарушении оттока желчи
Желтуха наблюдается при нарушении оттока желчи (обтурационная или механическая) из-за атрезии желчных путей, обтурации их камнем, сдавления желчных путей опухолевым процессом, увеличенными лимфатическими узлами или сгущением желчи. Механическое препятствие оттоку желчи приводит к застою и повышению давления желчи, расширению и разрыву желчных капилляров и поступлению желчи прямо в кровь или через лимфатические пути. Желтуха нарастает постепенно (за счет конъюгированного билирубина).
Холестаз: семейный доброкачественный возвратный, злокачественный семейный, чолестатический синдром, внутриклеточный холестаз
Семейный доброкачественный возвратный холестаз. В патогенезе его имеет значение гипоплазия лимфатических сосудов печени и других органов. Клинически заболевание проявляется признаками холестаза уже в неонатальном периоде. Затем холестаз самостоятельно уменьшается, рецидивирует у взрослых.
Злокачественный семейный холестаз проявляется на первом году жизни желтухой, которая самостоятельно разрешается через несколько недель или месяцев. Затем интенсивность желтухи нарастает, присоединяется сильный зуд. Печень и селезенка существенно увеличиваются.
Холестатический синдром может наблюдаться у детей при холангиохолитах, когда к признакам нарушения оттока желчи присоединяются симптомы бактериальной инфекции (лихорадка, лейкоцитоз, сдвиг лейкоцитарной формулы, повышенная СОЭ). У детей раннего возраста холангиохолит может быть осложнением тяжелых энтеритов, оперативных вмешательств на кишечнике, механических препятствий оттоку желчи.
При внутриклеточном холестазе в отличие от внепеченочного отсутствуют боли в области печени; расширение желчных ходов, увеличение органа незначительные.
источник